
When 9-year-old Jeremy Brown is in pain, it feels like he is being stabbed, while the pain experienced by Deborah Oliver, 40, is like a hundred simultaneous charley horses.
Brown, of Bridgeport, and Oliver, of New Haven, have sickle cell disease (SCD), a genetic blood disorder that causes excruciating pain, life-threatening complications and a shortened life expectancy. Almost one-half of sickle cell patients die in their 40s.
The disease affects some 100,000 Americans, about one in 365 African Americans and one out of 16,300 Hispanics; and in lesser numbers, people with Middle Eastern, Indian, Caribbean and Mediterranean ancestries. An estimated 2,000 people in Connecticut have SCD.
But the disease—discovered over 100 years ago—receives little research, funding and attention.
• Just two medications have been developed to treat the disease: hydroxyurea, approved in 1998; and Endari, approved in 2017.
• There is no national data registry for tracking the disease.
• Only four of the state’s 27 acute care hospitals have sickle cell treatment programs. And the last SCD awareness program by the Department of Public Health (DPH) was in 2007.
• A 2013 study in the journal Blood reported that cystic fibrosis, which affects 30,000 people nationally, receives seven to 11 times more funding per patient than sickle cell disease. The amyotrophic lateral sclerosis (ALS) challenge in 2014 raised more than $115 million for about 20,000 patients in the U.S. The bulk of the funds—$77 million—were allocated for research.
“I think it’s ignored because it’s predominantly a disease of inner-city African Americans,” said Dr. William Zempsky, a pain specialist at Connecticut Children’s Medical Center in Hartford, which treats children with SCD.

Derek Torrellas Photo.
Deborah Oliver at her home in New Haven. She was diagnosed with SCD at 4 years old.
The other treatment programs are at Yale New Haven and Bridgeport hospitals and UConn Health in Farmington. In total, they treat about half of the state residents with SCD, estimated Dr. Biree Andemariam, the UConn program director. There are no hard statistics on adults with SCD born before 1990, when the state started testing newborns for it. More than 660 babies were born with it from 1990 to 2017, according to DPH.
Andemariam said that about 1,000 patients get no care or go to emergency rooms when in crisis. She and Zempsky created an ER treatment protocol for hospitals without sickle cell programs but none of the Connecticut ERs they approached use it, she said.
Advocates say poor ER care has caused deaths due to lack of knowledge about SCD. Virginia Pertillar, executive director of Citizens for Quality Sickle Cell Care, said patients endure harmful, “unnecessarily long waits,” are accused of seeking opioids “to get high, not to relieve pain,” and are “mistreated or maltreated.”
“Quite honestly, because this primarily affects people of color in this country, some of the disparities in care and research dollars and pharmaceutical interests are intertwined with our country’s history of the marginalization experienced by people of color,” Andemariam said.
Dr. Gregory Buller, Bridgeport Hospital’s chief of medicine, said most SCD patients don’t have the income or clout to raise awareness, with many on disability or a limited work schedule. “If Warren Buffett had sickle cell disease, then the approach to it might be a whole lot different,” he said.
Treatment
In SCD, blood cells are sticky and deformed, causing clots, hampered blood flow, intense pain and, potentially, strokes, organ damage and breathing problems. Most adult patients have chronic pain. Children and adults have pain crises that erupt unpredictably and can last more than a week.
Bone marrow transplants are the only cure, but they’re risky and limited to very sick people who can find a donor match and who don’t have organ damage. Yale New Haven Children’s Hospital has done about a dozen successful transplants, said Dr. Farzana Pashankar, a hematologist-oncologist who specializes in SCD.
Pashankar said the hospital has expanded potential donors from siblings to also include parents and nonrelatives so more patients can be eligible for transplants.
University of Illinois Hospital doctors reported in April that they cured seven adults of SCD by using stem cells from family donors who previously would have been considered incompatible because their cells were only partial matches. One patient has died.
Dr. John D. Roberts, medical director of Yale New Haven’s adult sickle cell program, said because of the death, “what’s unclear is whether it was really successful.” He said that the results translate to a 12.5 percent death rate, “probably too high.”
In Paris, doctors reported that they have cured a boy with SCD using gene therapy. Scientists have called this development encouraging and promising but needing long-term follow up and more cases.
Treatments include opioids, transfusions, and the two SCD-targeted medications, hydroxyurea and Endari.
Jeremy Brown, a third-grader who wants to be an actor, has the most severe and debilitating form of SCD. Two years ago, he began taking hydroxyurea, a chemotherapy drug. He has been hospitalized once since. Before that, he was hospitalized at least monthly starting when he was 6 months old, said his mother, Tangi Small. Her husband lost a job after absences due to Jeremy’s hospitalizations. The couple and their four children became homeless, living in transitional housing for three years.
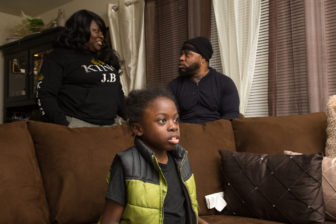
Derek Torrellas Photo.
Jeremy and his parents, Tangi Small and Jersino Brown, at their home.
Jeremy has endured intense pain in his arms and legs, fever, a distended stomach, bulging eyes and pneumonia. He can’t gain weight. Twice he couldn’t breathe on his own and was connected to a machine that removed some sickle cells and replaced them with normal ones. He has been prescribed morphine and oxycontin.
Oliver, a clinical technologist, has a different strain. She has pain crises about once a year, for which she is hospitalized and gets blood transfusions. She had her gall bladder removed and a hip replacement due to SCD complications. She limps from bone deterioration in her other hip. She is frequently online, searching for SCD advice from other patients. She was diagnosed when she had pneumonia at age 4.
“I’m not afraid to die,” Oliver said. “That’s not a fear. My concern is not fulfilling whatever my purpose is. I try to live each day as if it’s my last. I try to get the fullness of each day. I have no regrets.”
Connecticut SCD program directors said they are reducing hospitalizations with outpatient care, including individualized care plans, managing pain and reducing stress with psychiatric care and social work counseling, non-opioid medications, and self-management of opioid use, depending on need.
Roberts said that in 2015, advocates successfully lobbied to include SCD among the debilitating illnesses eligible for treatment with medical marijuana. It was not included in the 2012 state law.
The median life expectancy for SCD is 42 for men and 48 for women, according to Kathryn Britos-Swain, state sickle cell coordinator. Fewer children are dying, but adult mortality is not improving. A study by investigators at Johns Hopkins University School of Medicine showed it got 1 percent worse each year between 1979 and 2005.

Derek Torrellas Photo.
Jeremy Brown and his cousin, Denosh Billie, 12, color at home in Bridgeport. Denosh also has SCD.
“This is obviously a concern to families living with sickle cell disease and to sickle cell physicians,” Roberts said.
Small said she cried when her son was diagnosed “because I knew people who had it and passed early.” Four of her friends died from SCD at ages 17, 18, 21 and 32.
Research
Connecticut sickle cell research includes: Yale using synthetic marijuana to test marijuana’s effect on sickle cell pain; UConn studying quality of life, mental health, chronic pain, trauma and cell membranes; Connecticut Children’s working on a web-based tool to manage pain; and Bridgeport, Yale and UConn studying infections.
Pharmaceutical company interest in funding research has risen, but it is still difficult to get funding, Andemariam said, citing her inability to obtain funding to complete her work on SCD patients’ trauma.
Zempsky predicted better treatment options in the next decade as a result of new research. But, he said, “right now, the status quo is not very good.”
For information on sickle cell:
The National Health, Lung, and Blood Institute
Sickle Cell Disease Association of America, Southern Connecticut,
Citizens for Quality Sickle Cell Care
Yale New Haven Hospital’s sickle cell programs
Sickle Cell Infusion Center, Bridgeport Hospital
New England Sickle Cell Institute, UConn Health
Connecticut Children’s Medical Center
Connecticut Department of Public Health, sickle cell information
Children’s camp, parent retreats: Hole in the Wall Gang Camp
I am disturbed to see Dr. William Zempsky state that the disease is predominantly ignored because sickle cell is a disease of the inner-city African Americans !